
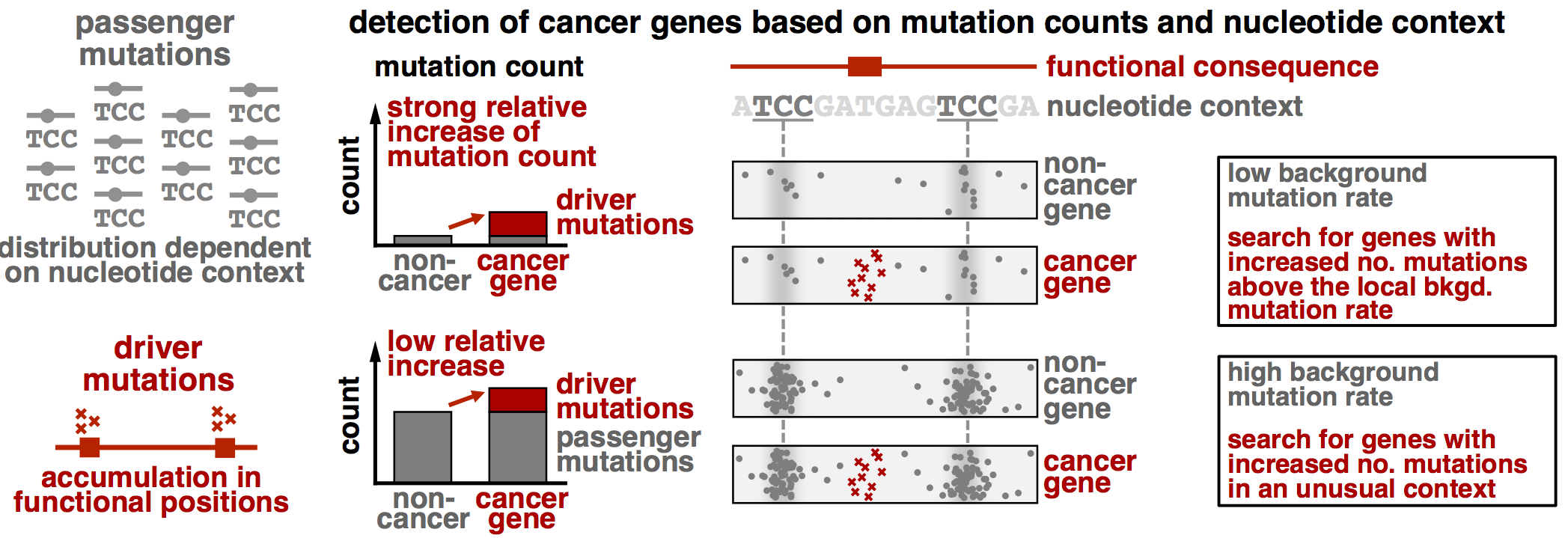
MutPanning is designed to detect rare cancer driver genes from aggregated whole-exome sequencing data. Most approaches detect cancer genes based on their mutational excess, i.e. they search for genes with an increased number of nonsynonymous mutations above the background mutation rate. MutPanning further accounts for the nucleotide context around mutations and searches for genes with an excess of mutations in unusual sequence contexts that deviate from the characteristic sequence context around passenger mutations.
Author: Felix Dietlein et al., Dana-Farber Cancer Institute and Broad Institute (algorithm), John Liefeld (module)
Contact:
Felix Dietlein (dietlein@broadinstitute.org) for algorithm questions.
Ted Liefeld (jliefeld@cloud.ucsd.edu) for module questions.
Algorithm Version: v2
Distributed under the BSD-3-Clause open source license. A copy of the license text is available at https://github.com/genepattern/docker-mutpanning/blob/develop/LICENSE.txt
| Name | Description |
|---|---|
| mutation file* |
The mutation file should be a tab-delimited standard *.maf format and contain the following columns: Hugo_Symbol (gene name), Chromosome, Start_Position ( |
| sample annotation file* |
The sample annotation file should be a tab-delimited *.txt file that contains at least two columns labeled Sample and Cohort. The Sample column contains unique sample name for each sample in the maf file. No duplicates allowed, all samples in the mutation file must be listed in the sample file. The cohort column contains the subcohort (e.g. cancer type) to which the sample belongs (case-sensitive). If you are unsure about the file format, you can download an exemplary sample file here. |
| min samples per cluster* | Minimum number of samples needed per nucleotide context cluster. Unless you are familiar with the MutPanning algorithm, we recommend running MutPanning with standard parameters (default value 3). |
| min mutations per cluster* | Minimum number of mutations needed per nucleotide context cluster. Unless you are familiar with the MutPanning algorithm, we recommend running MutPanning with standard parameters (default value 1000). |
| min samples Bayesian model* | Minimum number of samples needed to calibrate the Bayesian background model. Unless you are familiar with the MutPanning algorithm, we recommend running MutPanning with standard parameters (default value 100). |
| min mutations Bayesian model* | Minimum number of mutations needed to calibrate the Bayesian background model. Unless you are familiar with the MutPanning algorithm, we recommend running MutPanning with standard parameters (default value 5000).* - required |
mutation file (example mutation file)
This file lists the positions of all somatic mutations in your cohort. This file should follow the standard mutation annotation format. Several mutation callers (e.g. MuTect) report their somatic mutations in this file format. Each row corresponds to an individual mutation, which is annotated by the following colums.
Hugo_Symbol The nomenclature of the symbol in Hugo nomenclature (genenames.org).
Chromosome The chromosome, on which the mutation was found. Please use X and Y for the sex chromosomes and not 23 and 24.
Start_Position The start position of the mutation (Hg19).
End_Position The end position of the mutation (Hg19).
Strand The strand on which the mutation was detected (both 1/-1 or +/- nomenclature are fine).
Variant_Classification
Variant_Type The type of the mutation, such as single base substitution (SNP), insersions (INS) or deletions (DEL). Please use the standard nomenclature used in MAF files. If you are unsure about the file format, please see the exemplary files below.
Reference_Allele The nucleotide expected in the Hg19 reference genome. Please note that if this mutation does not with the in the Hg19 genome, this mutation is ignored.
Tumor_Seq_Allele1 The alternative nucleotide 1 found in the reads.
Tumor_Seq_Allele2 The alternative nucleotide 2 found in the reads. Typically, either of these nucleotides is the reference allele (allele A), whereas the other column contains the alternative read (allele B). Different callers handle differently how they assign these columns. MutPanning will first look in Tumor_Seq_Allele1 whether this equals the reference allele. If so, will take the change in the second Tumor_Seq_Allele column as alternative read.
Tumor_Sample_Barcode Th
sample.annotation.file (example sample annotation file)
This file organizes samples into subcohorts, e.g. cancer types. Samples in each subcohort are analyzed together for mutational significance. Each row in this file corresponds to an individual sample, which is annotated by the following columns.
Sample The same sample identifier as used in the mutation annotation file (case-sensitive). Note that these sample identifiers must be unique. Avoid special characters.
Cohort The cohort name, in which the samples should be analyzed together for significance. This is typically the cancer type, but you can also group your samples by other criteria (e.g., subtypes of cancer types or combine different cancer types together).
MutPanning.zip
The output of MutPanning is a zipped folder, which contains a mutational significance report for each subcohort (e.g. cancer type). If the cohort size is too small to calibrate the Bayesian background model (typically for cancer types with a very low background mutation rate, cf. parameters), the folder contains two analysis reports, based on the Bayesian model and a uniform background model.
Each significance report is a tab-delimited txt file, which lists genes in descending order according to their mutational significance. Each row corresponds to an individual gene, which is annotated by the following columns:
Name Gene name
TargetSize The nonsynonymous target size of the gene corrected for nucleotide context bias (sum of likelihood coefficients of nonsynonymous positions).
TargetSizeSyn The synonymous target size of the gene corrected for nucleotide context bias (sum of likelihood coefficients of synonymous positions).
Count Number of nonsynonymous mutations
CountSyn Number of synonymous mutations
Significance Mutational significance of the gene according to MutPanning. This p-value considers the excess of nonsynonymous mutations, the increased number of mutations in unusual nucleotide contexts, as well as insertions and deletions.
FDR The MutPanning p-value corrected for multiple hypothesis testing (q-value, Benjamini-Hochberg procedure).
If you are unsure about the file formats, you can download exemplary the following exemplary files:
Sample File
Mutation File
Task Type:
Tutorial
CPU Type:
Any
Operating System:
Docker
Language:
Java, Python
| Version | Release Date | Description |
|---|---|---|
| 2 | 2019-08-27 | Changing version number to 2.0 so that module and algorithm version numbers align |