p>The CopyNumberInferencePipeline is a method for analyzing Affymetrix SNP6.0 data files in GenePattern. The modules take Affymetrix SNP6 CEL files and process them in a pipeline that outputs genotype calls, copy number values, and copy number variant (CNV) regions. This page and the pipeline documentation can help you figure out how best to use this tool for your analysis. This pipeline includes a number of modules for data processing and analysis, with branches that allow for some additional analyses, depending on your input and desired output. Note that the segmented output of this pipeline can be used to run GISTIC.
The algorithms that constitute this pipeline were developed by the Cancer Genomics Computation Analysis group of the Broad Institute.
NOTE: If you have data from earlier Affymetrix SNP chips, you should use the SNPFileCreator module and look at the methodology suggested in the GenePattern "SNP Copy Number and Loss of Heterozygosity Estimation" protocol, available from the center pane of the main GenePattern page. Do not use the CopyNumberInferencePipeline.
Summary
The CopyNumberInferencePipeline accepts a set of raw SNP6 CEL files as input and generates segmented copy number calls for each sample. The copy number calls that are output are relative, and are normalized to make each sample appear to be diploid.
Workflow Overview
The general workflow of the CopyNumberInferencePipeline follows this outline:
- Calibrate signal intensities. (SNPFileCreator_SNP6)
- Compute genotype calls. (Birdseed)
- Convert signal intensities to copy number calls. (CopyNumberInference)
- Calculate copy number noise. (CopyNumberNoise)
- Reduce noise by removing outlier probes. (RemoveCopyNumberOutliers)
- Reduce noise further by subtracting out variation seen in a pre-defined set of normal samples. (TangentNormalization)
- Segment the copy number. (ParallelCBS)
- Calculate the number of segments in the genome and compare against a defined threshold to check for hyper-segmentation. (SegmentationCount)
NOTE: The pipeline is broken into two parts for ease of processing. For the specific workflow as it applies to the two parts of the pipeline, see this flowchart.
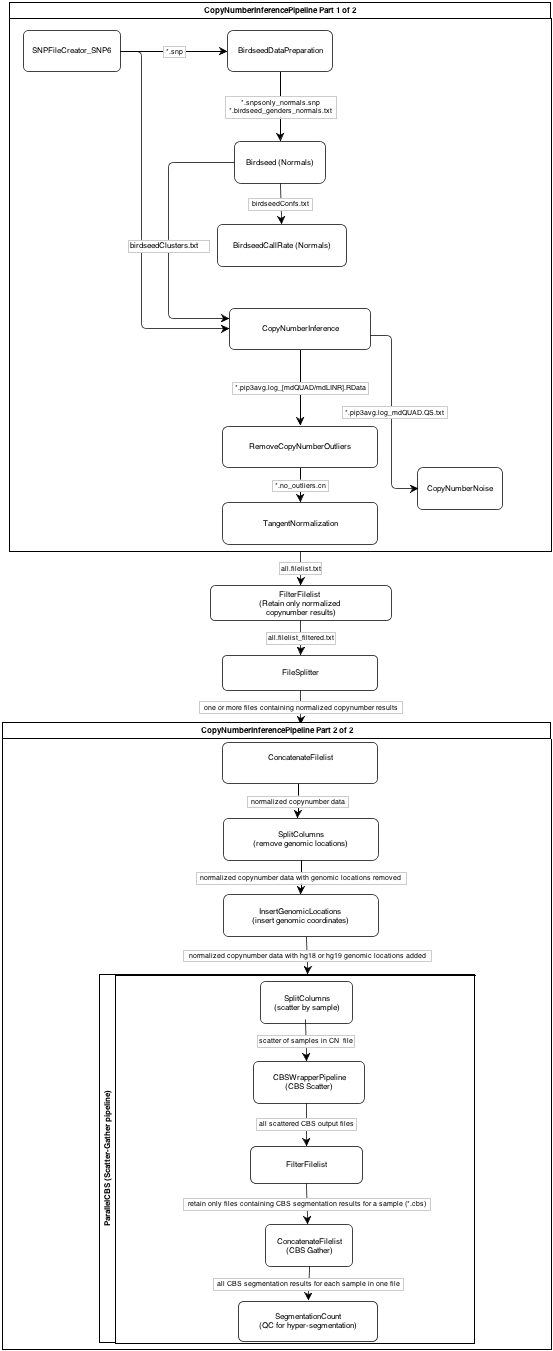{kind=link}
Input Requirements
Ideally all the SNP6 chips in the set (A) were processed as part of the same PCR batch and (B) should include 20 or more diploid normals within the PCR batch.
If your input consists only of normal samples, you should select the output with CNVs option when running the pipeline.
If your input is a set of tumor and normal samples, the algorithm will pass with a minimum of ten normal samples. Two considerations for normal samples are (1) that they are derived from true diploid cells, and (2) that they minimize batch effects as detailed below.
(1) You may prefer to use blood normal samples over tumor-adjacent tissue samples, as adjacent tissue are sometimes contaminated by tumors or early tumorigenesis events. For this reason, the authors of the pipeline typically use blood normals. Additionally, avoid cell lines as they are often no longer diploid. The normal samples should be unambiguously diploid; otherwise, the analysis can give misleading results.
(2) If these ten CEL files are random, the results come out noisier than if they are from the same batch. This is because the algorithm uses the normal samples to help calibrate out batch effects from the tumor samples. If you lack diploid normal samples from the same batch, use normal CEL files from samples that minimize batch effects. For example, normals should be from the same lab as the tumor samples, prepared using the same protocol, and if possible processed with the same batch of reagents, etc.
Due to memory constraints this pipeline is estimated to run with a maximum of about 200 CEL files.
Workflow Details
- Calibrate signal intensities (SNPFileCreator_SNP6)
This step adjusts the raw intensity values from the SNP6 array so that they can be compared with other arrays that were run within the same PCR batch. The distribution of the probes within each array is forced to follow a normal distribution, using quantile normalization, and this distribution is centered on a common value. In addition, the probes are reduced to a single intensity value for each unique probe sequence. The SNP6 array has only a single copy of each copy number probe; these are located at sites believed to be non-polymorphic. However, the SNP6 array has three replicates of the A allele and three replicates of the B allele for SNP probes. For the copy number probes, no additional work is needed, but for the SNP probes, the three replicates are merged using the median polish algorithm. See the SNPFileCreator_SNP6 module documentation for details.
- Compute genotype calls (Birdseed)
The normal samples in the batch are run through the Birdseed genotyping algorithm. In addition to providing genotype calls, this step also computes the cluster centers for the intensities of each probe for the AA, AB, and BB genotypes. By default, a set of prior samples also influence the cluster center locations. The normal samples are assumed to be diploid, and thus the cluster centers should reveal the expected intensity of zero, one, and two copies of each allele of the SNP probes.
- Convert signal intensities to copy number calls (CopyNumberInference)
This step converts intensity measurements into a copy number call. It uses different models for the copy number and SNP probes. For the SNP probes, copy number vs. intensity is modeled by a straight line connecting the zero and one copy cluster centers, as given by the Birdseed module. This calibrates out certain batch conditions. Copy number probes cannot calibrate out the batch in this way, so their model is based on a previous X-dosage experiment, where the number of X chromosomes in the cell line were independently measured by karyotyping. This model uses, in addition to the probe intensity, the GC content of the probe, the restriction fragment length, and the average intensity of the normals in the batch. Copy number calls from the SNP and copy number probe models are interleaved together to form a single set of copy number calls.
- Calculate copy number noise (CopyNumberNoise)
A noise QC metric is computed based on these copy number calls by examining the size of the step between probes that are adjacent in hg19 probe ordering. The threshold is selected based on examining its distribution, identifying the knee on a q-q plot that separates outliers from the main distribution.
- Reduce noise by removing outlier probes (RemoveCopyNumberOutliers)
The copy number calls are de-noised by removing probes that are outliers and replacing them with "NaN". Although probes that frequently perform poorly were already unconditionally removed, additional probes are identified which have radically different copy number calls than their hg19-adjacent neighbors. Four neighbors upstream and downstream are considered.
- Reduce noise further by subtracting out variation seen in a pre-defined set of normal samples (TangentNormalization)
The tangent normalization algorithm substantially further de-noises the signal by subtracting out variation that is also seen within a panel of normals. The default is to use a panel of more than 3000 blood normals from the Cancer Genome Atlas (TCGA), along with the normals that are in the current batch. In a tumor/normal sample set, a QC metric is computed on the normals in the current batch, to exclude those that have obvious broad copy number changes, as are sometimes found in tumor-adjacent normal tissue. Linear regression is used to select a linear combination of normals to subtract from each sample, excluding a fixed set of probes known to have a lot of germline variation. Probes in potential CNV regions do not have the normals subtracted from them, but are simply scaled to roughly match the scaling applied to the non-CNV regions. The list of excluded probes can be found on the properties page for TangentNormalization module.
- Segment the copy number (ParallelCBS)
In preparation for segmentation, the probes are sorted based on the order of the specified genome build, hg18 or hg19. And, for the ‘nocnv’ segmentations, a fixed set of probes are excluded (the same set that tangent normalization excludes due to frequently containing germline CNVs.)
The CBS segmentation algorithm identifies regions in the genome that, in spite of noise, probably have a uniform underlying copy number. It compresses the values from a set of adjacent probes into a single value for that interval. The intervals it generates are non-overlapping, and are usually abutting except when a NaN value happens to occur at a segment boundary. CBS can be run independently on either hg18 or hg19 builds, both including the germline CNVs and excluding them (nocnv). If you are using the data to look for somatic alterations, or using an algorithm such as GISTIC to identify somatic alterations that are statistically significant, you will get better results by using the nocnv segmentation results.
- Calculate the number of segments in the genome and compare against a defined threshold to check for hyper-segmentation (SegmentationCount)
Noisy samples exhibit hyper-segmentation, where CBS calls a huge number of segments that is unlikely to reflect the underlying biology. This QC metric counts the total number of segments in the genome, and compares it to a threshold. This threshold is set by examining a q-q plot of its log and identifying the knee, which distinguishes the tail from the main distribution.