Creating Input Files
When you run an analysis module, visualization module, or pipeline, GenePattern displays the parameters for the selected modules. Often, one or more of these parameters are input files, which must have a particular format; for example, you might need to supply a gct or res file. For more information about a particular file format, select it from the list at the right.
This section provides general information to help you create properly formatted files for GenePattern.
- Transforming Plain Text Files
- Creating GCT/RES Files
- Converting and Processing Files
- Converting CDT to GCT Files
Several video tutorials are also available:
- Converting Expression Data into a GenePattern Input File Format
- Converting Between MAGE-TAB and GenePattern Expression File Formats
- Converting Illumina Expression Data Into the GenePattern GCT format
- Importing Data from caArray to GenePattern
- Exporting Datasets from InsilicoDB to GenePattern
- Imputing Missing Values in GenePattern
Additional resources are available from our partnered site GenomeSpace. Their Convert file formats page lists supported file conversions, descriptions of file types, and an email to request new file format converters. Check their blog for newly added converters.
Transforming Plain Text Files
Although different GenePattern modules require different file formats, all of the files are plain text files. Plain text differs from rich text as described in Wikipedia, and these cannot be interchanged.
Data within files are parsed in various ways, e.g. tab-delimited, space-delimited, comma-separated, or one item per line. Most gene expression data is tab-delimited and you can open the file in a spreadsheet or database program that allows you to export the data into a plain text file. To transvert a plain text file to a specific format, e.g. GCT or CLS, the contents must conform to the attributes of the format as described and have the correct file extension. Required attributes include a specific delimiter, e.g. tab, specific usage for select cells, e.g. headings and numeric tallies, and/or unique sample and probe identifiers. File extensions appended to the end of a file are recognized by GenePattern modules, start with a period, and are typically three letters, e.g. dataset1.gct or dataset1.cls.
Multiple modules exist under Data Format Conversion and Preprocess & Utilities that transform plain text files to specific formats, e.g. Read_group_trackingToGct, help create supporting files from such expression files, e.g. ClsFileCreator, or process files in desired ways, e.g. UniquifyLabels and SelectFileMatrix.
Alternatively, you may manually transform files to format-specific plain text files using the following steps:
-
Open the data file in a text editor or spreadsheet editor.
- Keep in mind some spreadsheet programs convert gene symbols to dates, e.g. Excel converts MARCH1 to 1-Mar, as well as introduce other irreversible auto-formatting conversions as described in Zeeberg, et al (2004). Avoid these conversion by using Excel's Text Import Wizard, e.g. via File>Import. In the third step of the Text Import Wizard, highlight the column(s) and select Text as the Column data format instead of the default General.
- Use Excel's Text Import Wizard to convert space delimited files to spreadsheet format, i.e. select Delimiters>Space, and to split labels separated by a longbar (|) into two columns, i.e. select Delimiters>Other: and type | in the textbox.
-
Make the necessary changes that conform to file format attributes described on this page and save as a plain text file.
- Excel's Save As>Tab delimited text and Comma Separated Values save as plain text with TXT and CSV extensions, respectively.
- For a space-delimited file, open the CSV file with a text editor and find and replace each comma with a space.
- Mac's TextEdit defaults to rich text format. Change this for the current document by selecting Format>Make Plain Text, and for future documents by setting TextEdit>Preferences>Format to Plain text.
-
Change the file extension to the appropriate extension, e.g. GCT or CLS.
- In Mac OS, to change the file extension, right-click on the file>Get Info, expand Name & Extension section, uncheck Hide extension option, then change the extension in the box provided.
In turn, open these plain text format files using (1) the Open with function and selecting a text editor, (2) adding a .txt extension to the end and doubling-clicking, or (3) importing into a spreadsheet program, e.g. Excel.
Creating GCT/RES Files
GenePattern provides several modules for creating GCT and/or RES files from gene expression data from various sources. For information about these modules:
- Login to the public GenePattern server: http://cloud.genepattern.org/gp/.
If you do not have a GenePattern account, you can register on the login page. - Review the GCT and RES Files page of the GenePattern protocols.
Converting and Processing Files
The Modules page of the GenePattern web site provides a complete list of the modules and pipelines available from the Broad Institute. Modules in the Data Format Conversion category convert files from one format to another. Modules in the Preprocess & Utilities category provide methods for importing and working with data files.
Converting CDT to GCT Files
One common question from GenePattern users is how to convert a cdt file to a gct file. Following is a brief tutorial that walks you through this process by converting sample.cdt to sample.imputed.gct:
- Save the sample.cdt file to your local drive and open it in Microsoft Excel.
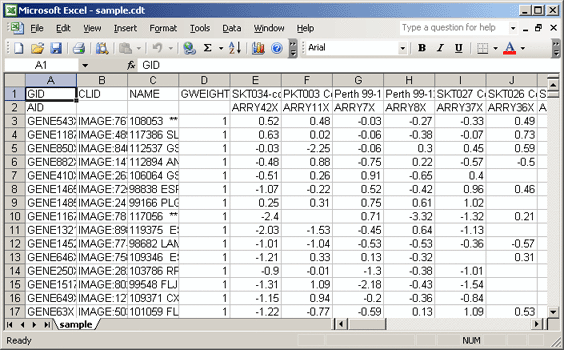
- Delete the CLID and GWEIGHT columns. The gct file format allows for only two columns of annotations.
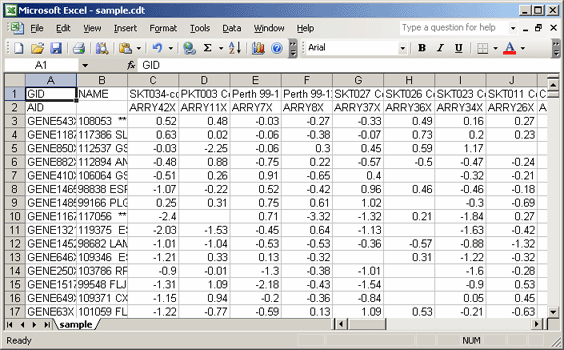
- Delete the second row, which contains array identifiers (AID). The gct file format allows for only one row of identifiers.
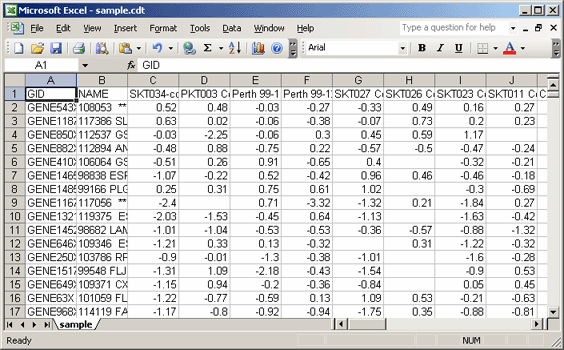
- Add two header rows at the top of the file:
- In the first row, first cell, enter: #1.2
- In the second row, first cell, enter the number of data rows: 1553
- In the second row, second cell, enter the number of data columns: 44
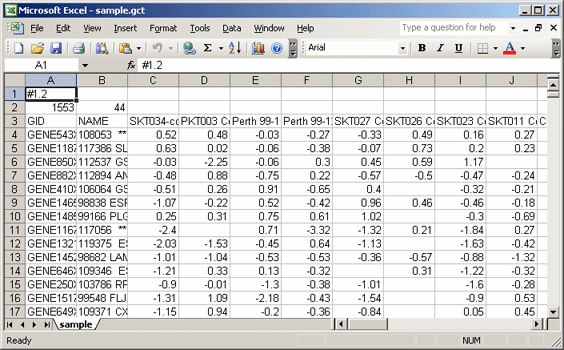
- Save the modified file as a text (tab delimited) file with the name sample.gct.
- Verify that your new .gct file matches the requirements of a gct file in GenePattern.
- Your original cdt file contained cells that were missing data. Most GenePattern modules require that all cells in a gct file contain data. Use the GenePattern analysis module ImputeMissingValues.KNN to add the missing data to your gct file. The module will take sample.gct as the input file, impute the missing data, and generate a sample.imputed.gct file.
ATR
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
ATR files will be created by the HierarchicalClustering module. This is a format defined at Stanford for their Hierarchical Clustering program. Note that the HierarchicalClustering module will also generate a CDT file.
The ATR (array tree) file records the order in which the arrays (columns) were joined during clustering.

CBS
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
CBS files are created by the CBS module. A CBS (circular binary segmentation) file is a tab-delimited text file with six columns: the sample id, the chromosome number, the map position of the start of the segment, the map position of the end of the segment, the number of markers in the segment, and the average value in the segment. The first row contains column headers and the second row onwards contains data values.
CDT
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
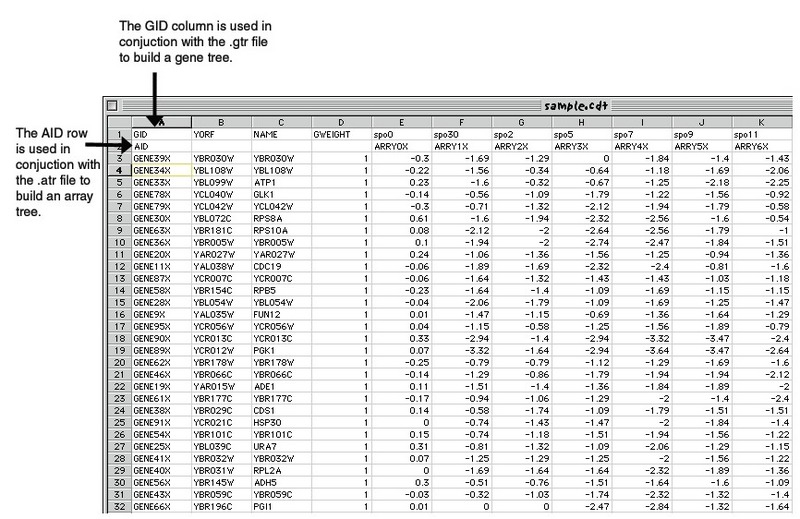
CDT files are created by the HierarchicalClustering module. This is a format defined at Stanford for their Hierarchical Clustering program. The CDT (clustered data table) file contains the original data, but reordered, to reflect the clustering. An additional column and/or row is added if clustering is performed on genes and/or arrays. The additional column/row contains a unique identifier for each row/column that is linked to the description of the tree structure in the GTR/ATR file also generated by the module:
- if you clustered by genes, a GTR file (gene tree)
- if you clustered by samples, an ATR file (array tree)
These tree files reflect the history of how the cluster was built, and can be used to reconstruct the tree(s).
CEL
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
Affymetrix image analysis software generates CEL files, which store information about the probes on a chip and the intensity values for the probes. GenePattern modules, such as ExpressionFileCreator and SNPFileCreator, take CEL files as input and generate data files that can be read by subsequent GenePattern modules.
CHIP
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
The CHIP file format contains annotation about a microarray (used with GSEA module). It lists the features (i.e probe sets) used in the microarray along with their mapping to gene symbols (when available). While this file is not used directly in the GSEA algorithm, it is used to annotate the output results and may also be used to collapse each probe set in the expression dataset to a single gene vector.
Chip annotation files can be specified in a tab-delimited file format (*.chip) or in a comma-separated file format (*.csv). The formats are identical other than the separation character (tab or comma). Typically, you use the tab-delimited (*.chip) file format.
The CHIP file format is organized as follows:
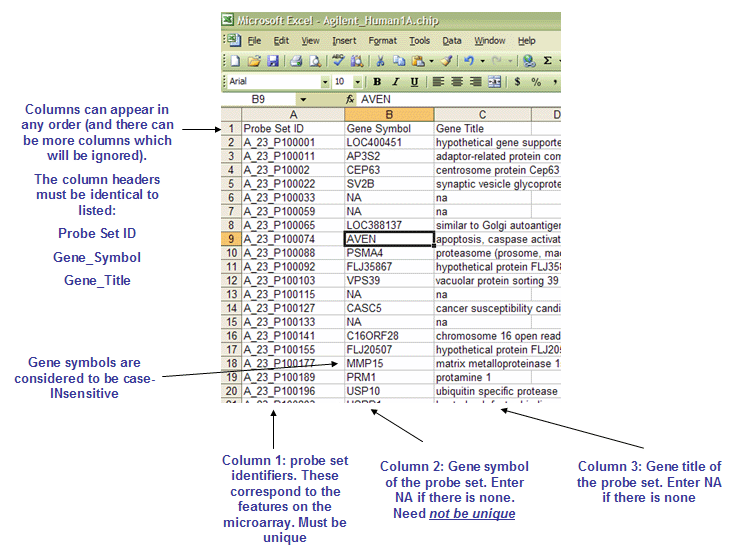
- The first line contains column headings that identify the content of each column in the remainder of the file. The file must contain three column headings:
- Probe Set ID
- Gene_Symbol
- Gene_Title
- For example:
Probe Set ID Gene_Symbol Gene_Title
- The rest of the data file contains data for each probe set ID used in the microarray.
- Line format:
(probe set id) (tab) (gene symbol) (tab) (gene title) - For example:
205699_at MAP2K6 mitogen-activated protein kinase kinase 6
- Line format:
Sample CHIP file: HG_U133A_annot.chip
CLM
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
The CLM file format is a tab-delimited file format that describes the samples in a zipped collection of CEL or IDAT files (used with the ExpressionFileCreator and IlluminaExpressionFileCreator modules, respectively).
Each row of the CLM format describes a CEL file in the zip file:
- Line format:
(CEL file name) (tab) (sample name) (tab) (class) - For example:
cat_a.CEL sample_cat_a tumor
- The first column contains the CEL file name (file extension is optional).
- The second column contains a sample name for the CEL file data.
- The third column contains a phenotype class name for the CEL file data.
Sample CLM file: sample.clm
CLS
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
The CLS file format defines phenotype (class or template) labels and associates each sample in the expression data with a label. It uses spaces or tabs to separate the fields. The CLS file format differs somewhat depending on whether you are defining categorical or continuous phenotypes:
- Categorical labels define discrete phenotypes, e.g. normal versus tumor.
- Continuous phenotypes are used for time series experiments or to define the profile of a gene of interest, e.g. gene neighbors.
Note: Most GenePattern modules are intended for use with categorical phenotypes. Therefore, unless the module documentation explicitly states otherwise, a CLS file should define categorical labels. GenePattern's ClsFileCreator module provides a guided wizard to create a CLS file from .gct or .res files.
Categorical labels
Categorical labels define discrete phenotypes (for example, normal vs tumor). For categorical labels, the CLS file format is organized as follows: 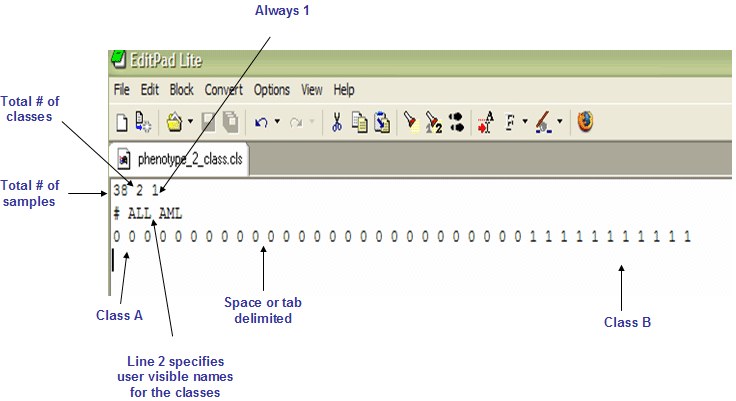
- The first line of a CLS file contains numbers indicating the number of samples and number of classes. The number of samples should correspond to the number of samples in the associated RES or GCT data file.
- Line format:
(number of samples) (space) (number of classes) (space) 1 - For example:
58 2 1
- Line format:
- The second line in a CLS file contains names for the class numbers. The line should begin with a pound sign (#) followed by a space.
- Line format:
# (space) (class 0 name) (space) (class 1 name) - For example:
# cured fatal/ref
- Line format:
- The third line contains a class label for each sample. The class labels are sequential numbers beginning with zero. The first label used (0) is assigned to the first class named on the second line; the second unique label (1) is assigned to the second class named; and so on. (NOTE: While most GenePattern modules adhere to this rule of 0 as the first class, some modules (such as GSEA) do not. Check the documentation for the module you are using if you are unsure.)
The number of class labels specified on this line should be the same as the number of samples specified in the first line. The number of unique class labels specified on this line should be the same as the number of classes specified in the first line.- Line format:
(sample 1 class) (space) (sample 2 class) (space) ... (sample N class) - For example:
0 0 ... 1
- Line format:
CLS file for sample RES file (categorical labels): all_aml_test.cls
Continuous labels
Continuous phenotypes are used for time series experiments or to define the profile of a gene of interest (gene neighbors). A CLS file that defines continuous labels can contain one or more labels. The following example shows a CLS file that defines two continuous labels:
#numeric #AFFX-BioB-5_st 206.0 31.0 252.0 -20.0 -169.0 -66.0 230.0 -23.0 67.0 173.0 -55.0 -20.0 469.0 -201.0 -117.0 -162.0 -5.0 -86.0 350.0 74.0 -215.0 193.0 506.0 183.0 350.0 113.0 -17.0 29.0 247.0 -131.0 358.0 561.0 24.0 524.0 167.0 -56.0 176.0 320.0 #AFFX-BioDn-5 75.0 142.0 32.0 109.0 -38.0 -80.0 62.0 39.0 196.0 -42.0 199.0 49.0 171.0 327.0 115.0 -71.0 85.0 80.0 270.0 182.0 208.0 -94.0 292.0 233.0 34.0 0.0 59.0 233.0 48.0 466.0 -7.0 -96.0 297.0 38.0 208.0 -15.0 30.0 357.0
- The first line contains the text "#numeric" which indicates that the file defines continuous labels.
- The remainder of the file defines the continuous phenotypes. For each phenotype:
- The first line defines the name of the phenotype; for example, #AFFX-BIOB-5_st.
- The second line contains a value for each sample in the .gct file. Typically, your word processor wraps the second line of the phenotype definition, as shown in the example.
For a continuous phenotype label, the values for the samples define the phenotype profile. The relative change in the values defines the relative distance between points in the phenotype profile. In the example shown above, the phenotype profile is the expression profile for a gene: the sample values for the two phenotype labels are gene expression values. For a time series experiment, you would choose sample values that define the desired expression profile. The example shown below assumes that you have five samples taken at 30 minute intervals. The first phenotype label defines a phenotype profile that shows steadily increasing gene expression; the second defines a profile that shows an initial peak and then gradual decrease:
#numeric #IncreasingProfle 30 60 90 120 150 #PeakProfle 5 20 15 10 5
CN
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
This is a tab-delimited file format that contains SNP copy numbers. It contains one row for each SNP and one column for each SNP array: the raw copy number value. It is organized as follows:
- The first line contains a list of labels identifying the SNP arrays.
- Line format:
SNP (tab) Chromosome (tab) PhysicalPosition (tab) (array_1_name) (tab) ... (array_N_name) - For example:
SNP (tab) Chromosome (tab) PhysicalPosition (tab) MYNAH_p_Affy_plate_9_Mapping250K_Sty_A01_49068 (tab) ... MYNAH_p_Affy_plate_9_Mapping250K_Sty_A01_49084
- Line format:
- The rest of the SNP file contains one row of data for each SNP.
- Line format:
(snp) (tab) (chromosome) (tab) (position) (tab) (array_1_cn) (tab) ... (array_N_cn) - For example:
SNP_A-4249904 (tab) 17 (tab) 41420045 (tab) 2.265 (tab) ... 1.735
- Line format:
Note: Sort the SNPs by chromosome and physical position (low to high). Most GenePattern modules, as well as many external tools, require sorted data.
Sample CN file: mynah.sorted.cn
FCS
Flow Cytometry Standard (FCS) files are used by GenePattern's numerous flow cytometry data analysis and data processing modules. The International Society for Advancement of Cytometry (ISAC) provides detailed resources outlining flow cytometry data file format standards, including for updated FCS formats, as well as example data transformations.
Check module documentation to see which versions of FCS files the module accepts. For example, the FLAME suite modules accept FCS v2.0, FCS v3.0, and TXT files.
In addition to analysis modules, GenePattern offers a number of modules that allow for FCS data manipulation that include CsvToFcs, FcsToCsv, ExtractFCSParameters, FCSNormalization, CompensateFCS, and PreviewFCS.
FPKM_tracking and Read_group_tracking
FPKM_tracking, Count_tracking, and Read_group_tracking files are output by the Cufflinks suite modules, which include Cufflinks and Cuffdiff. For more information on each of these and other Cufflinks suite file types, see the Cufflinks website. You can convert these to GCT format using Fpkm_trackingToGct or Read_group_trackingToGct modules.
FPKM_tracking files contain RNA-seq estimated expression values in Fragments Per Kilobase of transcript per Million mapped reads (FPKM) and use a generic format to output estimated expression values. Each FPKM tracking file has the following format:
| Column number | Column name | Example | Description |
| 1 | tracking_id | TCONS_00000001 | A unique identifier describing the object (gene, transcript, CDS, primary transcript) |
| 2 | class_code | = | The class_code attribute for the object, or "-" if not a transcript, or if class_code isn't present |
| 3 | nearest_ref_id | NM_008866.1 | The reference transcript to which the class code refers, if any |
| 4 | gene_id | NM_008866 | The gene_id(s) associated with the object |
| 5 | gene_short_name | Lypla1 | The gene_short_name(s) associated with the object |
| 6 | tss_id | TSS1 | The tss_id associated with the object, or "-" if not a transcript/primary transcript, or if tss_id isn't present |
| 7 | locus | chr1:4797771-4835363 | Genomic coordinates for easy browsing to the object |
| 8 | length | 2447 | The number of base pairs in the transcript, or '-' if not a transcript/primary transcript |
| 9 | coverage | 43.4279 | Estimate for the absolute depth of read coverage across the object |
| 10 | q0_FPKM | 8.01089 | FPKM of the object in sample 0 |
| 11 | q0_FPKM_lo | 7.03583 | the lower bound of the 95% confidence interval on the FPKM of the object in sample 0 |
| 12 | q0_FPKM_hi | 8.98595 | the upper bound of the 95% confidence interval on the FPKM of the object in sample 0 |
| 13 | q0_status | OK | Quantification status for the object in sample 0. Can be one of OK (deconvolution successful), LOWDATA (too complex or shallowly sequenced), HIDATA (too many fragments in locus), or FAIL, when an ill-conditioned covariance matrix or other numerical exception prevents deconvolution. |
| 14 | q1_FPKM | 8.55155 | FPKM of the object in sample 1 |
| 15 | q1_FPKM_lo | 7.77692 | the lower bound of the 95% confidence interval on the FPKM of the object in sample 0 |
| 16 | q1_FPKM_hi | 9.32617 | the upper bound of the 95% confidence interval on the FPKM of the object in sample 1 |
| 17 | q1_status | 9.32617 | the upper bound of the 95% confidence interval on the FPKM of the object in sample 1. Can be one of OK (deconvolution successful), LOWDATA (too complex or shallowly sequenced), HIDATA (too many fragments in locus), or FAIL, when an ill-conditioned covariance matrix or other numerical exception prevents deconvolution. |
| 3N + 12 | qN_FPKM | 7.34115 | FPKM of the object in sample N |
| 3N + 13 | qN_FPKM_lo | 6.33394 | the lower bound of the 95% confidence interval on the FPKM of the object in sample N |
| 3N + 14 | qN_FPKM_hi | 8.34836 | the upper bound of the 95% confidence interval on the FPKM of the object in sample N |
| 3N + 15 | qN_status | OK | Quantification status for the object in sample N. Can be one of OK (deconvolution successful), LOWDATA (too complex or shallowly sequenced), HIDATA (too many fragments in locus), or FAIL, when an ill-conditioned covariance matrix or other numerical exception prevents deconvolution. |
Cuffdiff calculates the expression and fragment count for each transcript, primary transcript, and gene in each replicate prior to differential expression calculations. The results are output in read group tracking files at the level of genes, isoforms, transcription start sites, and coding sequences. Each Read_group_tracking file has the following format:
| Column number | Column name | Example | Description |
| 1 | tracking_id | TCONS_00000001 | A unique identifier describing the object (gene, transcript, CDS, primary transcript) |
| 2 | condition | Fibroblasts | Name of the condition |
| 3 | replicate | 1 | Name of the replicate of the condition |
| 4 | raw_frags | 170.21 | The estimate number of (unscaled) fragments originating from the object in this replicate |
| 5 | internal_scaled_frags | 4905.63 | Estimated number of fragments originating from the object, after transforming to the internal common count scale (for comparison between replicates of this condition.) |
| 6 | external_scaled_frags | 99.21 | Estimated number of fragments originating from the object, after transforming to the external common count scale (for comparison between conditions) |
| 7 | FPKM | 201.334 | FPKM of this object in this replicate |
| 8 | effective_length | 5988.24 | The effective length of the object in this replicate. Currently a reserved, unreported field. |
| 9 | status | OK | Quantification status for the object. Can be one of OK (deconvolution successful), LOWDATA (too complex or shallowly sequenced), HIDATA (too many fragments in locus), or FAIL, when an ill-conditioned covariance matrix or other numerical exception prevents deconvolution. |
GCT
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
The GCT file format is a tab delimited file format that describes an expression dataset. The main differences between RES and GCT file formats are the RES file format (1) contains labels for each gene's absent (A) versus present (P) calls as generated by Affymetrix's GeneChip software and (2) does not allow missing expression values. Although the GCT file format allows missing values, only a few modules (such as CART, GSEA and HierarchicalClustering) can be run against an expression dataset that is missing values. Most modules do not allow missing expression values.
The GCT file is organized as follows:
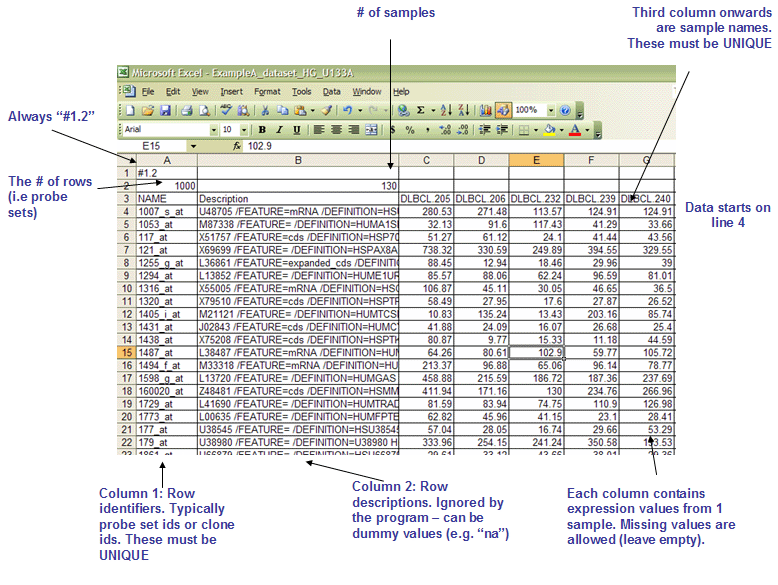
- The first line contains the version string and is always the same for this file format. Therefore, the first line must be as follows:
- #1.2
- The second line contains numbers indicating the size of the data table that is contained in the remainder of the file. Note that the name and description columns are not included in the number of data columns.
- Line format:
(# of data rows) (tab) (# of data columns) - For example:
7129 58
- Line format:
- The third line contains a list of identifiers for the samples associated with each of the columns in the remainder of the file.
- Line format:
Name (tab) Description (tab) (sample 1 name) (tab) (sample 2 name) (tab) ... (sample N name) - For example:
Name Description DLBC1_1 DLBC2_1 ... DLBC58_0
- Line format:
- The remainder of the data file contains data for each of the genes. There is one line for each gene and one column for each of the samples. The first two fields in the line contain name and descriptions for the genes (names and descriptions can contain spaces since fields are separated by tabs). The number of lines should agree with the number of data rows specified on line 2.
- Line format:
(gene name) (tab) (gene description) (tab) (col 1 data) (tab) (col 2 data) (tab) ... (col N data) - For example:
AFFX-BioB-5_at AFFX-BioB-5_at (endogenous control) -104 -152 -158 ... -44
- Line format:
Occasionally, GCT files are organized in a transposed structure where the columns represent genes and the rows represent samples. The user should take care to check the organization of the file to ensure that the correct preprocessing is performed on the file. See sample *.gct files that come with the distribution for complete examples of the format.
Sample GCT file: allaml.dataset.gct
GLAD
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
This is a tab-delimited file format that contains the output results of the GLAD module. The GLAD module, a SNP analysis module, runs the R package Gain and Loss analysis of DNA (GLAD), which detects altered regions in the genomic pattern. The GLAD file format is organized as follows:
- The first line contains a list of labels identifying the columns.
- Line format:
Sample (tab) Chromosome (tab) Start.bp (tab) End.bp (tab) Num.SNPs (tab) Seg.CN
- Line format:
- The rest of the file contains one row of data for each altered chromosomal region.
- Line format:
(sample) (tab) (chromosome) (tab) (startPosition) (tab) (endPosition) (tab) (numberOfSNPs) (tab) (regionCN) - For example:
MYNAH_p_Affy_plate_9_Mapping250K_Sty_A02_49084 (tab) 17 (tab) 41419603 (tab) 36581538 (tab) 6427 (tab) 2.06
- Line format:
Sample GLAD file: mynah.glad
GMT
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
The GMX and GMT file formats are tab-delimited file formats that describe gene sets (used with the GSEA module). In the GMX format, each column represents a gene set; in the GMT format, each row represents a gene set. The GMX format is convenient for storing a relatively small number of gene sets (<256) and is easier to edit. The GMT format is more convenient for storing larger databases of gene sets. The GMT format contains a row for each gene set:
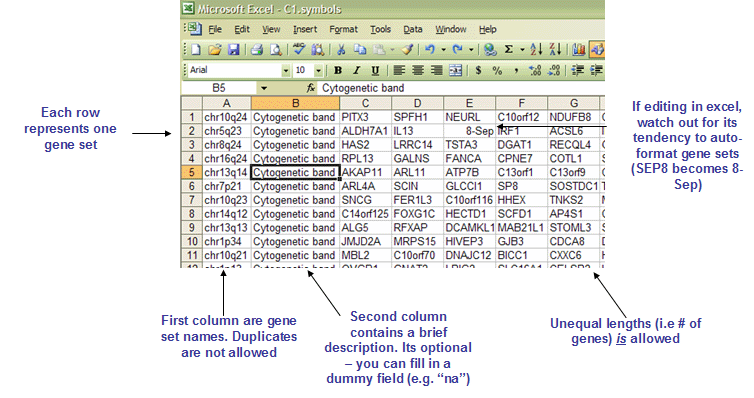
- Line format:
(gene set name) (tab) (description) (tab) (gene 1) (tab) (gene 2) (tab) ... (gene N) - For example:
GNF2_SPTA1 na ALS2CR3 KLF1 SLC6A8 ... CA1
- The first column contains the gene set name. Duplicate names are not allowed.
- The second column contains the gene set description. GSEA uses the description field to determine what hyperlink to provide in the report for the gene set description: if the description is na, GSEA provides a link to the named gene set in MSigDB; if the description is a URL, GSEA provides a link to that URL.
- The remaining columns list the genes in the gene set.
Sample GMT file: export_gnf.GENE_SYMBOL.gmt
GMX
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
The GMX and GMT file formats are tab-delimited file formats that describe gene sets (used with the GSEA module). In the GMX format, each column represents a gene set; in the GMT format, each row represents a gene set. The GMX format is convenient for storing a relatively small number of gene sets (<256) and is easier to edit. The GMT format is more convenient for storing larger databases of gene sets. The GMX format contains a column for each gene set:
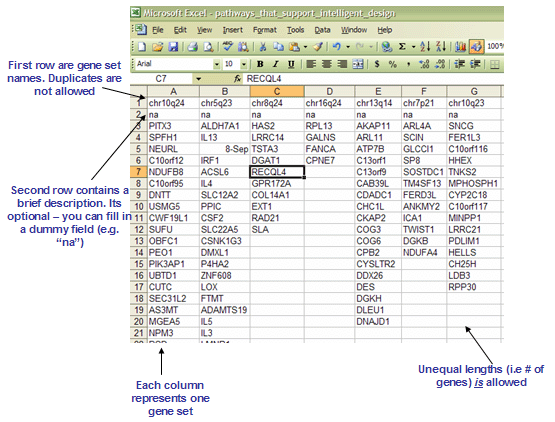
- Column format:
(gene set name) (tab) (description) (tab) (gene 1) (tab) (gene 2) (tab) ... (gene N) - For example:
GNF2_SPTA1 na ALS2CR3 KLF1 SLC6A8 ... CA1
- The first line contains the gene set name. Duplicate names are not allowed.
- The second line contains the gene set description. GSEA uses the description field to determine what hyperlink to provide in the report for the gene set description: if the description is na, GSEA provides a link to the named gene set in MSigDB; if the description is a URL, GSEA provides a link to that URL.
- The remaining lines list the genes in the gene set.
Sample GMX file: export_gnf.GENE_SYMBOL.gmx
GRP
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
The GRP file format contains a single gene set in a simple newline-delimited text format. Typically, you use the GMT or GMX file formats to create gene sets, rather than using the GRP file format. The GRP format contains a line for each gene, one gene per line. Lines that start with a pound sign (#) are ignored.
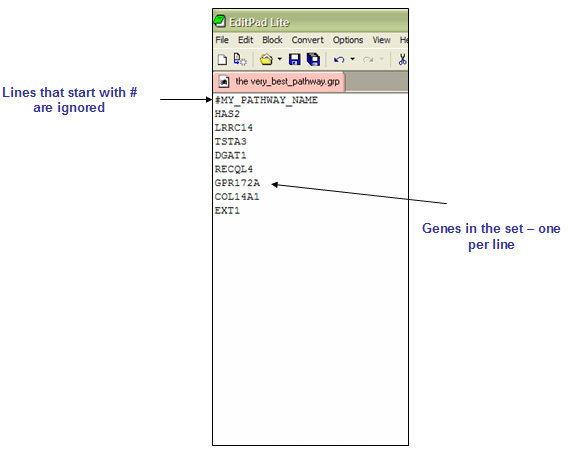
Sample GRP file: my_gene_set.grp
GTR
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
GTR files will be created by the HierarchicalClustering module. This is a format defined at Stanford for their Hierarchical Clustering program. Note that the HierarchicalClustering module will also generate a CDT file.
The GTR (gene tree) file records the order in which the genes (rows) were joined during clustering.
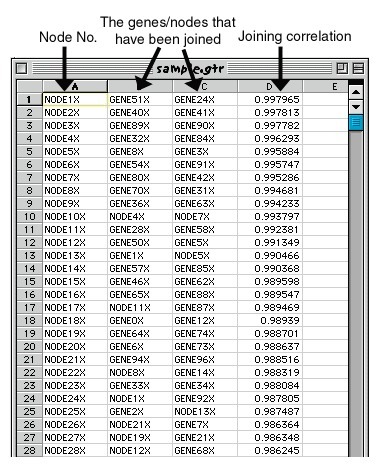
LOH
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
This is a tab-delimited file format that contains the output results of the LOH module. The LOH module, a SNP analysis module, detects loss of heterozigosity (LOH). The LOH file format is organized as follows:
- The first line contains a list of labels identifying the paired samples.
- Line format:
SNP (tab) Chromosome (tab) PhysicalPosition (tab) (pair_1_name) (tab) ... (pair_N_name) - For example:
SNP (tab) Chromosome (tab) PhysicalPosition (tab) SM-12VZ (tab) SM-12W1
- Line format:
- The rest of the SNP file contains one row of data for each probe.
- Line format:
(snp) (tab) (chromosome) (tab) (position) (tab) (pair_1_loh) (tab) ... (pair_N_loh) - For example:
SNP_A-1855068 (tab) 17 (tab) 41089766 (tab) R (tab) R
- L (LOH): AB in normal and A or B in tumor
- R (Retention): AB in both normal and tumor or No Call in normal and AB in tumor
- C (Conflict): A or B in normal and AB in tumor
- N (Non-informative call): A or B in normal, or No Call in normal or tumor
- Line format:
Sample LOH file: mynah.loh
MAF
The MutSigCV module utilizes Mutation Annotation Format (MAF) files. A MAF file is a tab-delimited plain-text file that notates mutation or variation against a reference genome. The file lists one mutation per row, gives information about the mutation in the columns, and has a header row that labels the columns. The National Cancer Institute's wiki on TCGA (The Cancer Genome Atlas) gives format specifications.
GenePattern modules utilizing MAF files require specific mutation information columns with specific column header labels. See the Input Files section of documentation for each module for the necessary columns. For example, MutSigCV MAF files must contain two nonstandard columns of effect and categ in addition to the columns gene and patient. A tab-delimited plain-text file containing only these four columns is sufficient to run the module.
MAGE-TAB and MAGE-ML
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
The MAGE-TAB and MAGE-ML file formats are defined by the Functional Genomics Data Society (FGED, formerly MGED) to create standards for the representation of microarray expression data to facilitate the exchange of microarray information between different data systems. More information about and sample files in the FGED-based formats are available here.
ODF
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
The Ouput Description Format (ODF) is similar to the RES or GCT file formats for datasets. The main difference is in the header. The body of data still contains the expression level values for each gene in each sample. Thus the main data block (after the header lines) is a matrix of values. The columns are defined by a name and optionally a description. The rows have a name (name of the gene for instance) and a description (description of the gene). The columns contain the expression values for each gene in a sample. If the first gene in the data block is a particular Tyrosine Kinase then each of the samples contained in each of the columns will have expressions values for that particular Tyrosine Kinase in the first row.
Note: This ODF format is specific to GenePattern. It is not an Open Document Format (ODF) for Office Applications as defined by the Organization for the Advancement of Structured Information Standards (OASIS).
ODF Header for Datasets
The following example shows the header lines of an ODF file. The first five lines are required.The line numbers are shown for easy reference, they should not be included in your file.
1. ODF 1.0
2. HeaderLines=7
3. Model= Dataset
4. DataLines= 3
5. COLUMN_TYPES: String String float float float *
6. COLUMN_DESCRIPTIONS: Sample from DFCI Sample from UK Sample from Children's
7. COLUMN_NAMES: Name Description Sample 1 Biopsy_2 Biopsy_4
8. RowNamesColumn=0
9. RowDescriptionsColumn=1
Lines 1 and 2 are required first and second lines. They must both be present in the header and be the first and second lines. They signify that this is an ODF formatted file (of type 1.0) and indicate the number of header lines that follow before the main data block (in this case 7 more). Line 3, required to be somewhere in the header of an ODF file, defines this ODF file as containing Dataset data. Line 4 is required somewhere in the header file. It indicates the number of data rows present in the data block. Line 5 is required somewhere in the header file for any ODF file that has a main data block. It defines the type of data in each column. Line 6 is a tab-delimited list of descriptions for each column. Line 7 is a tab-delimited list of names for the columns. Line 8 defines which column will have the row names, and Line 9 defines which column will contain the row descriptions.
Note: Following are a few notes about the ODF Header:
- The first element of each header line will be a key word. The keyword defines/describes what kind of meta data will be found on the rest of that line.
- A remark is a human readable comment that is skipped by the parser. This line starts with the "#" character and can contain any type of text since it is not parsed. Note that remarks are not counted as header lines and the user can insert them "by hand".
- The number of data lines can be quite large. For example, the U133A chip measures the expression values for about 20,000 human genes. A Dataset created from several samples using the U133A gene chip could have a large value for the DataLines tag.
- The COLUMN_NAMES:, COLUMN_DESCRIPTIONS:, and COLUMN_TYPES: lists must have the same number of elements. Also the number of elements must be equal to the number of columns in the main data block.
- The COLUMN_NAMES:, and COLUMN_DESCRIPTIONS: could be empty, that is simply contain the proper number of tabs but no text.
Main Data Block
The following example shows the first few lines of the main data block:
1000_at X60188 HSERK1 Human ERK1 mRNA 145.3 240.37823 158.66888
1001_at X60957 HSTIEMR Human tie mRNA 20.5 31.139397 14.053186
1002_f_at X65962 HSCP450 H.sapiens mRNA -9.6 118.06088 -8.287777
The main data block must be consistent with the header. The first COLUMN_NAMES element is "Name". This label is associated with the first column (values: 1000_at, 1001_at, and 1002_f_at). The second column's label is "Description" which is associated with the second column of the main data block. The next three columns are floating point numbers that represent the gene expression values for each of the samples.
Note: The first two columns are just text data, and next three columns only contain floating point values. This is consistent with the "String, String, float, float, float" elements in the COLUMN_TYPES: list.
Sample ODF file: all_aml_train.preprocessed0.odf
PCL
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
PCL files may be used as input for the GSEA module. This is a format defined at Stanford for their CDNA expression data. More information on this format is available here.
POL
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
The POL file format represents a Parameter-Ordered List. This format is a tab-delimited file with 4 columns, consisting of the following:
- A ranking (an integer corresponding to the rank of the feature)
- Unique feature identifier
- Feature description
- Distance metric (value upon which the rank position is based)
If you don't have a distance metric, you can use the rank in column 4 as well. Lines from a sample pol file are shown below:
0 X59798_at CCND1 Cyclin D1 0.0 1 S69272_s_at Cytoplasmic antiproteinase 465.5319538 2 U37012_at Cleavage and polyadenylation specificity factor 493.1997567 3 X69910_at P63 mRNA for transmembrane protein 493.5331802 4 U53347_at Neutral amino acid transporter B mRNA 515.3552173 5 M80899_at AHNAK AHNAK nucleoprotein (desmoyokin) 539.9990741
RES
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
The RES file format is a tab delimited file format that describes an expression dataset. The main differences between RES and GCT file formats are the RES file format (1) contains labels for each gene's absent (A) versus present (P) calls as generated by Affymetrix's GeneChip software and (2) does not allow missing expression values. The file is organized as follows:
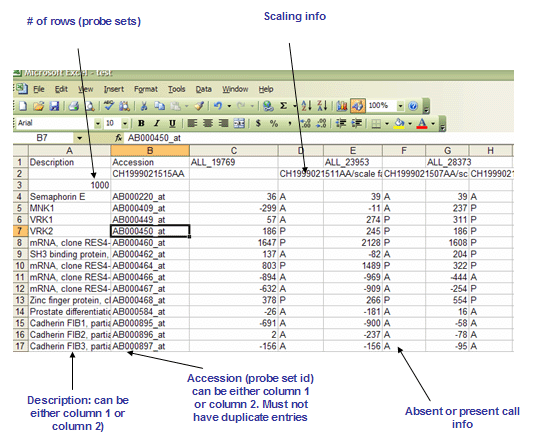
- The first line contains a list of labels identifying the samples associated with each of the columns in the remainder of the file. Two tabs (\t\t) separate the sample identifier labels because each sample contains two data values (an expression value and a present/marginal/absent call).
- Line format:
Description (tab) Accession (tab) (sample 1 name) (tab) (tab) (sample 2 name) (tab) (tab) ... (sample N name) - For example:
Description Accession DLBC1_1 DLBC2_1 ... DLBC58_0
- Line format:
- The second line contains a list of sample descriptions. Currently, GenePattern ignores these descriptions.
- Line format:
(tab) (sample 1 description) (tab) (tab) (sample 2 description) (tab) (tab) ... (sample N description) - For example, our RES file creation tool places the sample data file name and scale factors in this row:
MG2000062219AA MG2000062256AA/scale factor=1.2172 ... MG2000062211AA/scale factor=1.1214
- Line format:
- The third line contains a number indicating the number of rows in the data table that is contained in the remainder of the file. Note that the name and description columns are not included in the number of data columns.
- Line format:
(# of data rows) - For example:
7129
- Line format:
- The rest of the data file contains data for each of the genes. There is one row for each gene and two columns for each of the samples. The first two fields in the row contain the description and name for each of the genes (names and descriptions can contain spaces since fields are separated by tabs). The description field is optional but the tab following it is not. Each sample has two pieces of data associated with it: an expression value and an associated Absent/Marginal/Present (A/M/P) call. The A/M/P calls are generated by microarray scanning software (such as Affymetrix's GeneChip software) and are an indication of the confidence in the measured expression value. Currently, GenePattern ignores the Absent/Marginal/Present call.
- Line format:
(gene description) (tab) (gene name) (tab) (sample 1 data) (tab) (sample 1 A/P call) (tab) (sample 2 data) (tab) (sample 2 A/P call) (tab) ... (sample N data) (tab) (sample N A/P call) - For example:
AFFX-BioB-5_at (endogenous control) AFFX-BioB-5_at -104 A -152 A ... -44 A
- Line format:
Sample RES file: all_aml_test.res
Sample Information File
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
The sample information file is a tab-delimited format that describes a set of SNP arrays. The column labels in the first row define the information provided for each array; each subsequent row describes one SNP array. The sample information file is organized as follows:
- The first line contains the column labels. A sample information file can contain any number of columns and the column labels are arbitrary. However, SNP modules may require specific labels, as discussed below.
- Line format:
Label-1 (tab) Label-2 (tab) ... Label-n - For example:
Array (tab) Sample (tab) Type (tab) Ploidy(numeric) (tab) Gender (tab) Paired (tab) Platform
- Line format:
- The remainder of the sample information file contains a line of information for each SNP sample. Where data is unavailable, columns may be empty.
- Line format:
Col-1- data (tab) Col-2-data (tab) ... Col-N-data - For example:
S004274N_250S_123005 (tab) S004274N (tab) Normal (tab) 2 (tab) (tab) (tab) 250K_Sty
- Line format:
A sample information file can contain any number of columns and the column labels are arbitrary. A SNP analysis module, however, may require a sample information file to include specific column labels. For example, the SNP module CopyNumberDivideByNormals requires a sample information file that includes two columns, Sample and Ploidy(numeric). Following is a list of commonly used column labels:
- Array: Identifier for the SNP array.
- Sample: Identifier for the biological sample used to generate the SNP array data.
- Type: Brief description of the biological sample.
- Ploidy(numeric): Integer value, where ploidy=2 indicates a normal sample.
- Gender: Identifier that indicates the gender of the biological sample donor. For a sample from a male donor, Gender=M; from a female donor, Gender=F.
- Paired: Value that identifies normal-target pairs. For the normal sample, Paired=Yes; for the target sample, Paired is set to the sample name of the paired normal sample.
- Platform: SNP chip used to generate the array.
Note: When a SNP module requires a sample information file to include specific column labels, the module documentation lists the required column labels. Specify required column labels exactly: they are case-sensitive and space-sensitive.
Sample .txt file: 250K_Sampleinfofile.txt
Creating a Sample Information File
The following steps outline how to copy exactly sample identifiers from Excel data and tranpose them from horizonal to vertical.
- In Excel, Select entire row containing sample names and Copy. Open a new workbook, Paste Special>Transpose.
- If starting from a RES file, to remove blank rows, Select relevant column(s), then click Edit>Go To>Special button>Blanks option and click OK. Blank rows will be selected. Choose Edit>Delete>Entire row option and click OK.
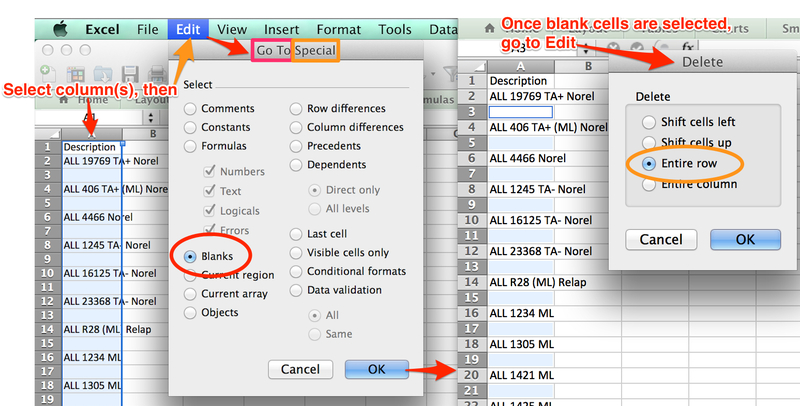
- Label row headings exactly as specified for module, fill in cells, and save as tab delimited text (.txt). For example, ComBat module labels first three cells of Row 1: “Array”, “Sample”, and “Batch”.
SNP
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
This is a tab-delimited file format that contains SNP array data. The SNPFileCreator module can create non-allele-specific SNP files or allele-specific SNP files. A SNP file contains one row for each SNP and two or three columns for each SNP array. Non-allele-specific files contain two columns for each SNP array: the intensity value and the call. Allele-specific files contain three columns for each SNP array: the intensity value for allele A, the intensity value for allele B, and the call. Note: Not all SNP modules accept allele-specific SNP files.
The first line of a SNP file contains a list of labels identifying the SNP arrays. The rest of the SNP file contains one row of data for each probe. Each row contains the probe intensity values and the SNP calls generated by the SNP microarray scanning software, such as Affymetrix's GeneChip software, as shown in the examples below.
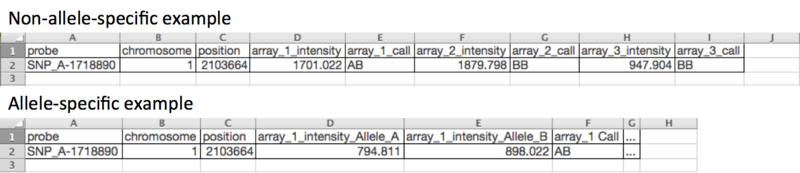
- Non-allele-specific files contain two columns per sample.
- Line format:
SNP (tab) Chromosome (tab) PhysicalPosition (tab) array_1_name (tab) array_1_name Call (tab)...array_N_name (tab) array_N_name Call ... - Example header row:
SNP (tab) Chromosome (tab) PhysicalPosition (tab) 100H_primary_GBM_101N (tab) 100H_primary_GBM_101N Call (tab) 100H_primary_GBM_56 (tab) 100H_primary_GBM_56 Call
- Line format:
- Allele-specific files contain three columns per sample--an intensity value for each of the two alleles and a call.
- Line format:
SNP (tab) Chromosome (tab) PhysicalPosition (tab) array_1_name_Allele_A (tab) array_1_name_Allele_B (tab) array_1_name Call ... - Example header row:
SNP (tab) Chromosome (tab) PhysicalPosition (tab) 1100H_primary_GBM_101N_Allele_A (tab) 100H_primary_GBM_101N_Allele_B (tab) 100H_primary_GBM_101N Call
- Line format:
Sample SNP file: gistic_subset.snp, gistic_subset_allele_specific.snp
TXT
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
The TXT format is a tab delimited file format that describes an expression dataset. It can be used with the GSEA module and is organized as follows:
- The first line contains the labels Name and Description followed by the identifiers for each sample in the dataset. The Description is optional.
- Line format:
Name(tab)Description(tab)(sample 1 name)(tab)(sample 2 name) (tab) ... (sample N name) - Example:
Name Description DLBC1_1 DLBC2_1 ... DLBC58_0
- Line format:
- The remainder of the file contains data for each of the genes. There is one line for each gene. Each line contains the gene name, gene description, and a value for each sample in the dataset. If the first line contains the Description label, include a description for each gene. If the first line does not contain the Description label, do not include descriptions for any gene. Gene names and descriptions can contain spaces since fields are separated by tabs.
- Line format:
(gene name) (tab) (gene description) (tab) (col 1 data) (tab) (col 2 data) (tab) ... (col N data) - Example:
AFFX-BioB-5_at AFFX-BioB-5_at (endogenous control) -104 -152 -158 ... -44
- Line format:
XCN
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
This is a tab-delimited file format that is similar to the SNP file format, but contains SNP copy numbers rather than SNP intensity values. It contains one row for each SNP and two columns for each SNP array: the raw copy number value and the call value. It is organized as follows:
- The first line contains a list of labels identifying the SNP arrays.
- Line format:
SNP (tab) Chromosome (tab) PhysicalPosition (tab) (array_1_name) (tab) (array_1_name) Call (tab) ... (array_N_name) (tab) (array_N_name) Call - For example:
SNP (tab) Chromosome (tab) PhysicalPosition (tab) MYNAH_p_Affy_plate_9_Mapping250K_Sty_A01_49068 (tab) MYNAH_p_Affy_plate_9_Mapping250K_Sty_A01_49068 Call (tab) ... MYNAH_p_Affy_plate_9_Mapping250K_Sty_A01_49084 (tab) MYNAH_p_Affy_plate_9_Mapping250K_Sty_A01_49084 Call
- Line format:
- The rest of the SNP file contains one row of data for each probe, including the raw copy number value and the SNP calls generated by SNP microarray scanning software (such as Affymetrix's GeneChip software). Some modules, such as the SNPViewer module, require the data sorted by chromosome and physical position.
- Line format:
(snp) (tab) (chromosome) (tab) (position) (tab) (array_1_cn) (tab) (array_1_call) (tab) ... (array_N_cn) (tab) (array_N_call) - For example:
SNP_A-4249904 (tab) 17 (tab) 41420045 (tab) 2.265 (tab) AB (tab) ... 1.735 (tab) AA
- Line format:
Sample SNP file: mynah.sorted.xcn
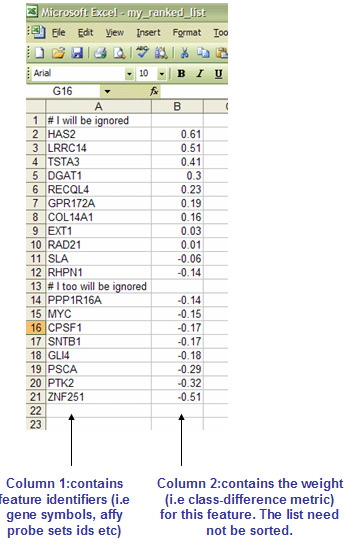
RNK
Note: If you are using Excel to edit GenePattern files, be sure to save the file as a tab-delimited text file and supply the correct file extension. You can specify the file name in quotes to prevent Excel from appending .txt to the file name. Also, note that Excel's auto-formatting can introduce errors in gene names, as described in Zeeberg, et al (2004).
The RNK file contains a single, rank ordered gene list (not gene set) in a simple newline-delimited text format. It is used when you have a pre-ordered ranked list that you want to analyze with GSEA. For instance, you might have used your favorite tTest-like statistic to produce a ranked ordered gene list from your dataset which you now want to test for enrichment. Order of lines does not matter. It is important, however, that the second column will have numeric values - they will be used to rank order genes by GSEA.